
В первой статье мы делились полезной информацией и ссылками, которые пригодятся для работы с биоинформатическими данными. Советуем сначала прочитать ее, если вы пропустили.
Дисклеймер
Работа с генетическими данными проводится на Unix системах (Linux, macOS), так как некоторые команды и ПО недоступны на Windows. Поэтому для пользователей Windows одним из самых простых решений будет арендовать виртуальную машину с Linux.
Все описанные ниже операции выполняются в командной строке — терминале. Перед началом выполнения узнайте, как работать в терминале под управлением вашей ОС и использовать команды, так как некоторые из них могут потенциально навредить ОС и вашим данным.
Необходимое ПО
Мы собрали образ виртуальной машины (ВМ) со всем необходимым ПО на Яндекс.Облаке. Зарегистрируйтесь в Яндекс.Облаке, в личном кабинете в разделе Compute Cloud нажмите «Создать ВМ». В качестве публичного образа выберите из каталога «Атлас Анализ Данных 1000 Геномов».
Конфигурация ВМ: 100% 2vCPU, 8GB RAM, 20GB HDD. При создании ВМ необходимо ввести платежные данные, но со счета ничего не спишется. Стартового и дополнительного гранта по кодовому слову достаточно, чтобы работать c ВМ и образом от Атласа до 31 декабря 2019 года бесплатно. Чтобы получить грант на прохождение задач, отправьте службе поддержки Яндекс.Облака кодовое слово «АТЛАС».
Примечание: грант действителен для новых пользователей Яндекс.Облака, которые завели аккаунт с 18 декабря 2019 года или для тех, у кого еще действует пробный период и имеется стартовый грант. Кодовое слово «АТЛАС» действует только один раз.
Предварительно создайте ssh ключ на локальном компьютере, с которого вы планируете подключаться к ВМ:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100 Не забудьте скопировать содержимое ~/.ssh/yandex-cloud.pub файла в соответствующее окно при создании ВМ.
Если вы хотите установить ПО на свой компьютер, ниже есть вся информация по установке. Если вы решили воспользоваться Яндекс.Облаком — создайте ВМ и переходите к следующему разделу.
Plink
Plink — программный пакет для манипуляций с генетическими данными и широкогеномного поиска ассоциаций (GWAS). Его разработал генетик Шон Перселл (Shaun Purcell). С помощью Plink с 2008 года были выполнены сотни GWAS’ов по всему миру, результаты лучших из которых Атлас использует как источник данных для алгоритмов расчета рисков заболеваний.
Plink предлагает набор инструментов для хранения и конвертации данных генотипирования и поиска по ним. Также Plink позволяет производить статистическую обработку, анализ неравновесного сцепления (linkage disequilibrium, LD), анализ идентичности по происхождению (identity by descent, IBD) и по состоянию (identity by state, IBS), популяционную стратификацию и тесты на эпистаз — взаимодействие нескольких генетических вариаций между собой.
IBD и IBS используются для анализа популяционного состава и определения родства.
Пример эпистаза — вариации rs7412 и rs429358 в гене APOE, определенное сочетание вариантов которых резко повышает риск развития болезни Альцгеймера, тогда как каждый вариант по отдельности вносит лишь незначительный вклад в риск.
Скачайте стабильную версию Plink с официального сайта.
BCFtools
BCFtools — набор утилит для манипулирования генетическими данными в формате VCF и его бинарном аналоге BCF. Список возможных применений пакета BCFtools включает аннотацию, фильтрацию, слияние и разделение VCF/BCF файлов, нахождение их пересечения, индексирование, выборочный поиск, сортировку, подсчет статистики и т.п.
Для установки выполните:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools # The following is optional: # autoheader && autoconf && ./configure --enable-libgsl --enable-perl-filters makeПодробнее процесс установки описан здесь.
KING
Пакет KING (Kinship-based INference for Gwas) используется в популяционных исследованиях при работе с данными полногеномного поиска ассоциаций для определения родственных связей в исследуемых данных. В этой задаче KING поможет определить степень родства нескольких образцов из проекта «1000 Геномов».
Скачать можно здесь. Для решения задач пригодится руководство к KING по ссылке.
Почти все ошибки, которые могут возникнуть в процессе работы с инструментами, описаны на Stackoverflow или его биоинформатическом аналоге — Biostars.
Используемые данные
Для руководства мы используем открытые данные проекта «1000 Геномов». Для анализа мы выбрали 10 образцов с информацией о генотипах около 85 миллионов вариаций, полученных путем анализа данных NGS с выравниванием на версию референсного генома GRCh37. Родственные связи и популяции образцов указаны на Рисунке 1.

Рисунок 1. Родословная используемых в VCF образцов. Квадрат соответствует мужскому полу, круг — женскому. Пунктирная линия означает неустановленное родство второго порядка.
Взять на заметку
Формат VCF позволяет хранить информацию о поле человека в виде одного числа, если эта информация была известна при генерации VCF. Выглядит это так: поле GT (genotype, генотип) для записей с Х хромосомы содержит одно численное значение, которое соответствует одной аллели, для мужчин и два — для женщин. Если информация о биологическом поле секвенируемого образца отсутствует, то поле GT будет по умолчанию содержать два числовых значения (выделены красным на рисунке 2).
В используемых в данном руководстве VCF файлах Y-хромосома исключена, но и наличие хромосомы Y в VCF файле далеко не всегда говорит о том, что она действительно есть у секвенируемого образца. Это происходит из-за псевдоаутосомных регионов (PARs), которые идентичны для Х и Y хромосом и расположены на их концах.
Разные хромосомы в норме не имеют длинных идентичных (гомологичных) участков, однако хромосомы Х и Y обладают такими участками длиной по несколько миллионов пар оснований в самом начале (PAR1) и конце (PAR2). Поэтому при анализе данных NGS у мужчин в PAR регионах обнаруживаются два аллеля (по одному с каждой половой хромосомы), а у женщин могут появиться генотипы в PAR регионах Y хромосомы, хотя на самом деле это генотипы с их X-хромосомы.
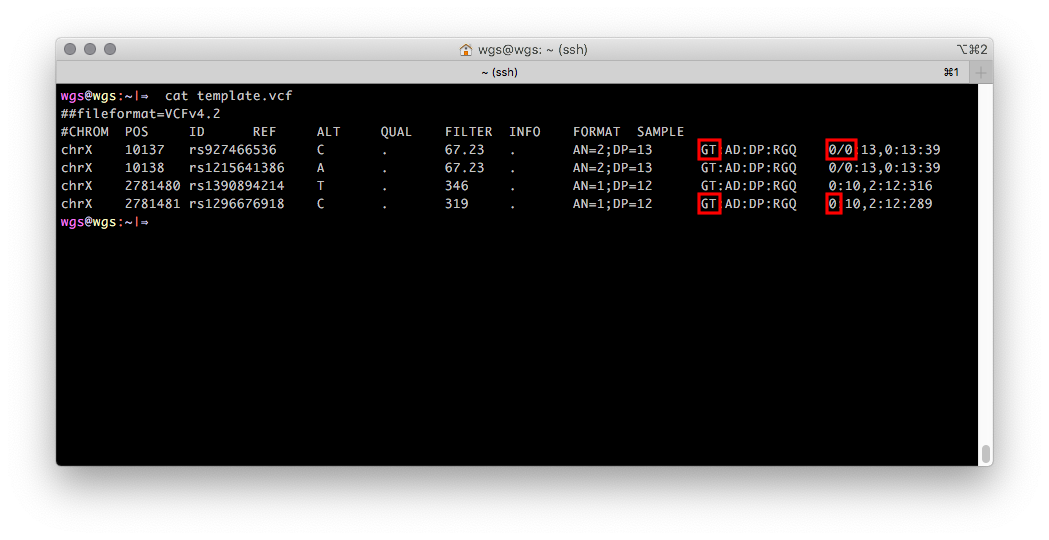
Рисунок 2. VCF файл с генотипами с Х хромосомы мужчины из региона PAR1 (первые две записи) и непсевдоаутосомного региона (последние две записи).
Образовательный блок
Генетический пол — это набор половых хромосом, соответствующих проявлению первичных и вторичных половых признаков по мужскому или женскому типу. В норме мужчины имеют одну хромосому Х и одну хромосому Y, в то время как женщины имеют две хромосомы X. При различных нарушениях образования половых клеток, яйцеклеток и сперматозоидов, у родителей может родиться ребенок с отличным набором половых хромосом, что часто приводит к нарушениям развития первичных и вторичных половых признаков.Две самых частых хромосомных половых аномалии — это синдром Тёрнера (набор хромосом X0, то есть всего одна X-хромосома) и синдром Клайнфельтера (набор хромосом XXY).
Аллель — один или более нуклеотид, который находится в какой-либо позиции генома и имеет альтернативный вариант. Понятие используют при описании генотипов. Различают референсные аллели и альтернативные. Все они хранятся в VCF файле в полях REF и ALT, соответственно.
Определяем пол
Tutorial содержит VCF файл, необходимый для выполнения руководства, папка Test — для выполнения самостоятельных заданий. В папке Technical содержится два файла со списком идентификаторов генетических вариаций: rsids_for_subsetting.txt используется в руководстве и заданиях для самостоятельного выполнения, external_interpretation_rsids.txt может понадобиться в будущем при приобретении полногеномного секвенирования в Атлас для загрузки данных генотипирования в сторонние сервисы. Папка Tools содержит помимо прочего два скрипта, используемые в заданиях 2 и 3.
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh В каталоге /home на ВМ Яндекс.Облака будет создана папка, название которой соответствует имени пользователя, указанному на этапе создания ВМ. Скопируйте всё из каталога /home/ubuntu в свой каталог через следующие команды:
cd ~ cp -r /home/ubuntu/* ./
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh Распакуйте atlas_wgs_contest.tar.gz архив командой
tar -xvzf atlas_wgs_contest.tar.gz
VCF файлы для выполнения заданий в неархивированном виде занимают около 19 гигабайт каждый, поэтому для экономии места мы рекомендуем работать только с архивами. Все перечисленные выше программы уже умеют работать со сжатыми данными VCF. Дополнительно ничего делать не нужно.
Чтобы определить пол субъекта, нужно посмотреть на генотипы на Х хромосоме и исключить регионы PAR1 и PAR2, расположенные в ее начале и конце. Это интервалы позиций 60001–2699520 и 154931044–155260560 в GRCh37 версии генома. Если генотип будет содержать одно численное обозначение — это мужской биологический пол, иначе — женский. Нужно иметь в виду, что обозначение пола в VCF файле зависит от наличия информации о биологическом поле при генерации VCF, поэтому использовать данный подход можно не всегда.
Используйте следующую команду для каждого из образцов в датасете. Подставьте идентификатор образца после аргумента -s:
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz): # перейдите в нужную вам директорию bcftools view -O v -H -s HG00731 -r chrX:2699521-154931043 CEI.1kg.2019.demo.vcf.gz | head При выполнении команд вы увидите часть содержимого VCF файла для указанного идентификатора образца. Параметр -r chrX:2699521-154931043 в BCFtools ограничивает просмотр содержимого файла до региона хромосомы Х с позиции 2699521 до позиции 154931043 (регион non-PAR на Рисунке 3). Эти границы исключают ненужные в данном случае псевдоаутосомные регионы (PAR1 и PAR2). По численным значениям в поле GT определите пол каждого образца.
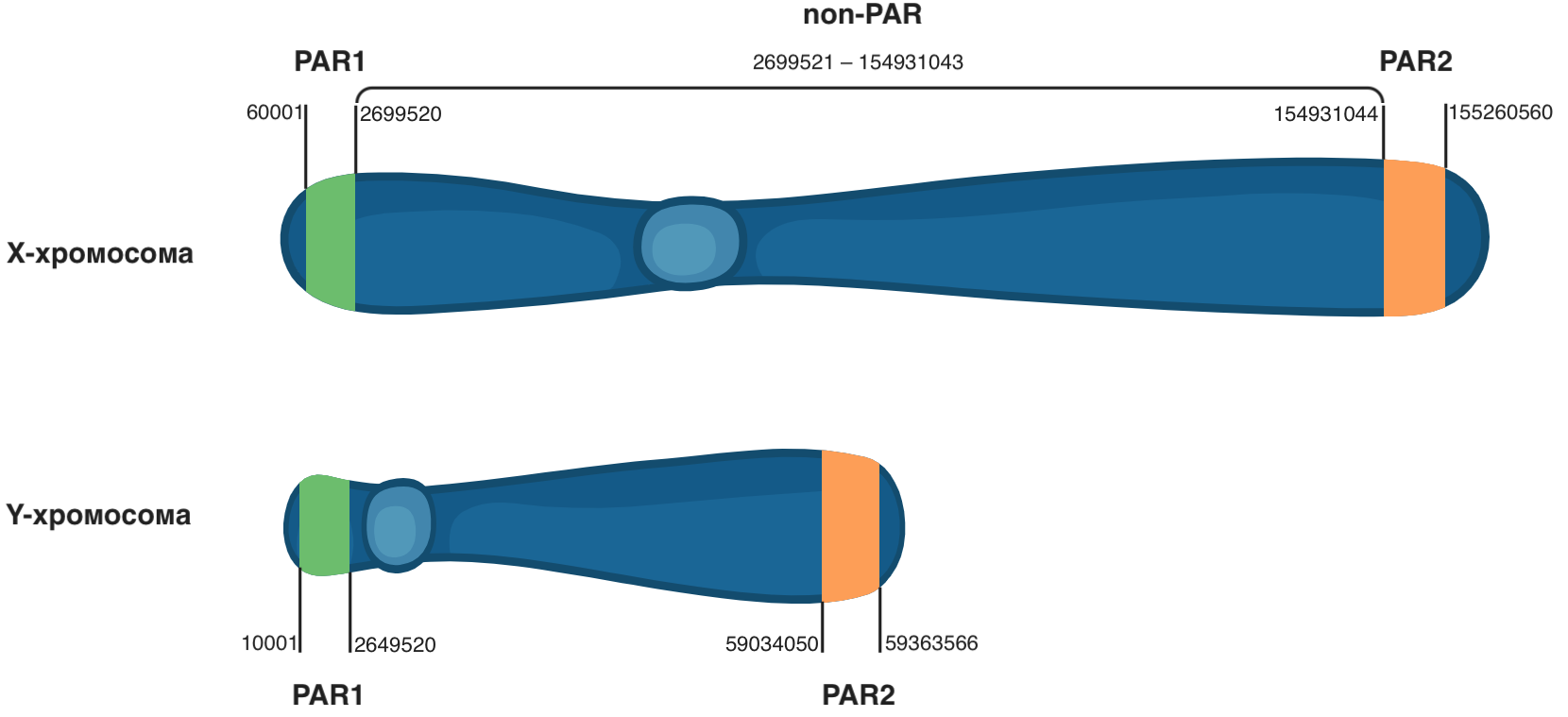
Рисунок 3. Расположение псевдоаутосомных регионов PAR1 и PAR2 на половых хромосомах.
Посмотреть список всех идентификаторов образцов в VCF файле можно на Рисунке 1 или в последней строке шапки VCF файла. Они будут перечислены после наименования колонки FORMAT:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT HG00731 HG00732 HG02026 HG02025 HG02250 HG02373 HG00733 HG02024 HG02377 HG02381Истинный пол данных образцов также указан на Рисунке 1.
Определяем родство
Для определения родства нам нужно попарно сравнить генетические данные всех образцов. Делать это по полному геному сложно: VCF файл в таком случае занимает десятки гигабайт. Используемый нами VCF занимает всего около 2 гигабайт, но мы все равно отфильтруем его по списку идентификаторов генетических вариаций (rsIDs), генотипируемых на чипах от компании Illumina: GSA v1, GSA v2, HumanOmniExpress v1.0, HumanOmniExpress v1.3, InfiniumExome v1.1 и InfiniumOmniExpressExome v1.4. Это наиболее популярные чипы в коммерческом генотипировании.
Мы собрали список всех идентификаторов генетических вариаций с этих чипов в отдельный файл со списком rsIDs. Он содержит 1,4 миллиона идентификаторов. Для фильтрации VCF файла выполните следующую команду:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gzПри каждом использовании BCFtools и других пакетов для работы с VCF файлами в шапку файла добавляется история предыдущих команд. Независимо от способа фильтрации VCF файла и выполненных ранее команд, можно проверить целостность и идентичность основного содержимого VCF подсчетом хеш-суммы:
# перейдите в нужную вам директорию gunzip -c CEI.1kg.2019.demo.subset.vcf.gz | grep -v "^#" > CEI.1kg.2019.demo.subset.body.vcf md5sum --tag CEI.1kg.2019.demo.subset.body.vcf # Вывод последней команды должен совпадать с d609eb330908d4eb5feb9f1860fd508b Команда gunzip -c производит декомпрессию файла с выводом его содержимого в stdout, из которого далее удаляются строки шапки VCF файла, начинающиеся с # (поэтому используется команда grep -v "^#"). Шапка удаляется для того, чтобы сравнивать целостность только самих генетических данных, а не метаданных о том, какие инструменты и когда были использованы для работы с этим VCF файлом.
В случае совпадения хеш-суммы можно двигаться дальше и конвертировать VCF во внутренний формат Plink (по умолчанию формат Plink — это три файла с расширениями bed, bim и fam). В этих файлах остается только генотип, хромосома, позиция и некоторые другие данные, а остальное — отсеивается. С таким форматом гораздо проще работать и решать различные задачи, не требующие дополнительной информации из VCF. Например проводить GWAS.
# Перейдите в нужную директорию plink --vcf CEI.1kg.2019.demo.subset.vcf.gz --make-bed --out CEI.1kg.2019.demo.subset Эта команда создаст в папке три файла:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.fam
Определить попарное родство можно для всех 10 образцов. Используем следующую команду для анализа Plink файлов:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis Просмотрите файл CEI.1kg.2019.demo.subset.kinship_analysis.kin0 и обратите внимание на колонку Kinship, в которой содержатся kinship коэффициенты для пар образцов, указанных соответственно в ID1 и ID2.
Сравните полученные вами коэффициенты в файле CEI.1kg.2019.demo.subset.kinship_analysis.kin0 для всех пар образцов с родословной, представленной на Рисунке 1 (пунктирная линия соответствует родству второго порядка, однако точные данные о родстве отсутствуют, т.е. это могут быть двоюродные брат и сестра, тетя/племянник или дядя/племянница). Попробуйте самостоятельно сделать вывод о том, какие значения kinship коэффициентов могут соответствовать родству первого и второго порядка.
Первое задание конкурса
Используя тестовый датасет из 12 образцов Data/Test/CEI.1kg.2019.test.vcf.gz, составьте их родословную, руководствуясь результатами определения пола и kinship анализа. Образцы, которые по результатам анализа не находятся в родственных связях с кем-либо, запишите рядом, не соединяя их линией с другими образцами. Родословная может быть составлена в стилистике, аналогичной Рисунку 1, однако это остается на ваше усмотрение. Мужчины обозначаются квадратом, женщины — кругом, брак — горизонтальной линией, ребенок — вертикальной линией, несколько детей — горизонтальным разветвлением вертикальной линии (в виде буквы П). Подробнее про эти обозначения можно почитать здесь.
Как мы писали выше, kinship коэффициенты не могут однозначно охарактеризовать родство того или иного порядка: одинаковые kinship коэффициенты получаются при сравнении пар родитель — ребенок и брат — сестра (родство первого порядка). При невозможности установить характер родственной связи, укажите любую из возможных. Обратите внимание, что образцы в тестовом датасете имеют отличные от используемых в датасете для обучения идентификаторы.
Ответы присылайте на почту wgs@atlas.ru до 26 декабря до 23:59. Еще две задачи будут скоро опубликованы, а итоговые результаты по задачам появятся 28 декабря. Победитель получит тест Полный геном, а второе и третье места — генетический тест «Атлас». Также будут специальные призы от Яндекс.Облако. Бывшие и настоящие сотрудники Атласа в конкурсе не участвуют 😉
ссылка на оригинал статьи https://habr.com/ru/company/atlasbiomed/blog/480954/
Добавить комментарий