Чуть ранее я написал статью Систематика прокариот — дальние родственники, где сообщил о грубых результатах и методе их получения. Он несколько не классический, но вполне укладывается в научную парадигму. Достаточно «жесткий» диалог с Davidov, который имел место быть в этой статье, может создать впечатление проблематичности метода о котором я говорю. Но мы потом сели и спокойно обсудили, и подвели некоторые итоги. Суть диалога представляет некоторый интерес и я его вначале частично опубликую.
А затем хочу продемонстрировать один наглядный пример построения дерева «происхождения видов» с помощью моего подхода (назовем его «детерминированный подход»). По сути метод можно обобщить, и тогда он не будет относится только к филогенетики и его можно использовать в других областях, когда нужно граф превратить в дерево, выкинув слабые связи.
Проблематика в диалогах
Диалог хоть и с коррекцией получился длинный. Но я бы рекомендовал его прочесть — он показывает разницу между моими взглядами и классическими взглядами в филогенетике. Главное противоречие у нас получилось в том, что считается, что нельзя найти более древние виды среди тех которые сейчас секвенировали, т.к. они современники (братья друг другу). Опровержение я попытаюсь дать в следующем разделе. В диалоге также видна разница между моим подходом (который я вкратце опишу в следующем разделе) и классическим. Если мой подход детерминированный и точный, то классический — это вероятностный. А уже из-за разных методов у нас и получаются разные противоречия в воззрениях — отсюда собственно и главное противоречие о нахождении более древних видов. На вероятностной основе это действительно нельзя никаким образом увидеть, но на детерминированной можно (правда с ограничениями, из-за недостаточности данных).
davydov: давайте с постановки задачи, чтобы потом не было сложностей.
я: ок
davydov: задачка такая: возможность восстанавливать эволюционные соотношения между отдельными видами бактерий. так?
я: термин эволюционные соотношения мне не нравится, под него можно подсунуть не то что надо
davydov: ок. давайте конкретнее тогда
я: Если говорить на бытовом уровне, то нужно понять как произошел организм, мог ли он произойти без того, чтобы до это не существал бы другой организм. В простейшем случае имеем организм А и организм Б, надо сказать какой из них предокdavydov: погодите, довольно странная задачка. Как более ли менее устроена эволюция? (очень упрощенно) есть некий универсальный общий предок — LUCA (last universal common ancestor) он себе живет и живет. тут бац, у него появились два потомка один начал менять в одну сторону, другой — в другую, прошло 2 миллиарда лет, и предка не осталось.
я: Изложу свою версию. Вначале есть множество РНК-нитей, они могут в разных комбинациях агрегироваться в один организм — так появляется первое поколение предков
davydov: допустим. РНК мир.
я: далее, они обмениваются различными РНК-нитями, которые эволюционируют в сторону чаще усложнения и отбираются в соответствии с пригодностью по функциональности. Этот начальный момент важен, потому, что сейчас суть эволюции не меняется. Дальше я согласен, что есть предки, которые затем умирают, но оставляют в своем геноме артефакты, по этим артефактам мы и можем судить, какие организмы более древние по своему составу
davydov: что значит более древние?
я: это означает, что например нить РНК достаточно консервативна и существовала в предках, и теперь её можно видеть в текущих видах без изменений, или с минимумом изменений
davydov: то есть древний означает «близкий к предковому состоянию», так?
я: да
davydov: хорошо. считается, что термин древний — не очень хороший термин, т.к. подразумевает время (а оно для всех ныне живущих одно и то же).
я: в том то и дело — нам важно именно время, точнее чтобы мы могли судить о этом времениdavydov: какое именно время?
я: Ну скажем время образования данной конкретной тРНК или белка
davydov: мы про виды сейчас.
я: да, но виды — это агрегаты РНК и белков (генетического материала), в виде может содержаться более древние РНК и более молодые, они образовались не в одно время
davydov: хорошо. то есть вас интересует, как давно появилась та или иная молекула, понял.
Но. Предположим, что вся эволюция генома — это мутации + дупликации + перестановки.
тогда получается, что все рнк произошли в момент t0.я: ок, но утверждение, что «все рнк произошли в момент t0» мне не понятно
davydov: ну дело в том, что у нас нету почти никакой информации про мир до LUCA. поэтому все склонны изучать эволюцию уже после появления luca, я условно называю этот момент t0.
я: Хорошо, но тогда точнее сказать, что к моменту t0 существовало определенное множество РНК
davydov: ну хорошо, но просто это ещё совсем не было похоже на современный организм.davydov: я сейчас скажу, с чем я совсем не согласен, мы потом попытаемся разобраться, прав я или нет. Почти все молекулы произошли так: (1) дупликация (2) дивергенция (то есть расхождение двух прежде идентичных копий). У LUCA согласно современным данным было всего лишь 29 белков, и наверное штук 30 других генов. Все остальные белки и гены возникли в результате геномных перестроек (в т.ч. дупликаций) и дивергенции
я: пока все ок
davydov: когда в результате дивергенции появляется что-то разумное (как с миоглобинами человека), оно попадает сначала под положительный (организмы с этим лучше выживают), а потом иногда под отрицательный (организмы без этого умирают) отбор, когда что-то находится под сильным отрицательным отбором (без этого легко сдохнуть), оно становится консервативным. пока все сходится с вами?
я: ага
davydov: отлично. так вот, большая часть молекул братья в плане происхождения. Был момент, когда они были мусорной днк, а до этого копией какого-то гена, а до этого ещё чем-то. А если все они братья, то вопрос о том, кто произошел раньше не имеет смысла, потому что у всех генов в сущности один предковый ген, который стал как-то там в водичке дуплицироваться, а потом дивергировать. Какой вопрос имеет смысл? — Это вопрос о том, в какой момент времени существовал предок кишечной палочки и обезьяны или вопрос о том, когда появились первые гены похожие на миоглобин. То есть последний (очень существенное слово) общий предок современных миоглобинов.я: тут уже пошло что-то не понятное, попробую раскрутить. Скажем рРНК вначале существовала в примитивном виде, и рибосома работала но хуже, далее рРНК начала усложняться и это помогло рибосоме лучше работать. Так как же не имеет смысла какой вид имела рРНК вначале, а какой потом? И главное в какое время произошла положительная мутация?
davydov: положительные мутации происходили постоянно и отрицательные (но они не закреплялись) мы можем зафиксировать появление нового белка, например. Но не в том смысле появление, что его не было раньше,
а в том смысле, что у всех ныне живых организмов этот белок — потомок того самого
я: и что? не важно когда происходили изменения?
davydov: дело не в том, что не важно. дело в том, что вы этого не увидите.
я: мы можем зафиксировать каждую положительную мутацию и с какого состояния она произошла, это разнообразие есть в видах, и их нужно лишь правильно выстроить
davydov: вот допустим перед luca у нас была простая ррнк. потом 3 млрд лет назад произошла положительная мутация
а luca возник допустим 2 млрд лет назад (это неверно на самом деле, но допустим). Значит у всех потомков luca эта положительная мутация будет. так? а если она будет у всех, то вы не знаете, когда она появилась.
я: нет она будет не у всех, а только у тех, которые близки к luca
davydov: даже если есть те, которые ближе к luca, чем другие (что отдельный вопрос), то почему у тех, кто дальше, её уже нет? почему её не будет у тех кто дальше от luca?
я: Нет их дальше, потому что происходит дальнейшие положительные мутации
davydov: а почему у тех, кто «близко» тоже не происходят положительные мутации? их заморозили? 🙂
я: потому что мутации происходят не в отдельной функциональной единице (РНК, белок) а на всем множестве ДНК… и да там могут начать положительно изменяться другие части, но отдельные функциональные единицы будут заморожены
davydov: почему у одних «далеких» видов за n млрд лет произошло много мутаций, а у «близких» мало?
я: почему много/мало? одинаково, только в разных местах раз, и два потому что были не положительные мутации, а отрицательные и это не закреплялось в разных местах ДНК в целом
davydov: в общем идея в том, что «близких» к luca нет. а есть те, которые стали эволюционировать в одну сторону. есть те, которые стали в другую.
я: скажем вероятность положительной мутации тРНК достаточно мала. Если меняется нуклеотид в консервативной части, то будет отрицательная мутация — и для этой тРНК очень сложно найти положительную мутацию, поэтому виды начинаю дивергенцию, и по отдельным элементам они будут ближе к luca, а по другим нет.
davydov: согласен с последним. то есть вы сейчас сказали, что есть консервативные молекулы и неконсервативные.
мне кажется, у нас опять путаница. есть консервативные (похожие на luca) молекулы, но нету консервативных (похожих на luca) видов. правильно?
я: похоже все так
davydov: отлично. мы все ближе, ладно, завтра о задаче.
я: до завтра. хотя о степени консервативности видов можно говорить, как о совокупности наличия более близких к luca консервативных молекул. Вот видимо в чем разница у нас.
я: последнию мою фразу вчера видел: «хотя о степени консервативности видов можно говорить, как о совокупности наличия более близких к luca консервативных молекул. Вот видимо в чем разница у нас.»?
davydov: да, с этим я согласен. то есть если мы можем восстановить «предковое состояние» по большому числу генов (что само по себе задача довольно непростая), то для каждого конкретного вида мы сможем определить, насколько он близок к этому предковому состояния. я думаю, что большой разницы между разными видами не будет, но определенно есть, для кого эволюция шла чуть быстрее, есть те, для кого чуть медленнее. Интуитивно подозреваю, что полученная величина будет очень хорошо коррелировать с длиной каждой конкретной ветви (от корня) для каждого конкретного вида.я: мы хотели поговорить о задаче!? Я говорю о идентичных тРНК — это такие тРНК, которые по своей последовательности полностью совпадают и не имеют мутаций в разных видах, родах и т.д. до типов и царств
davydov: постановка задачи — это тот биологический вопрос, на который мы ищем ответ.
я: Так вот на основании эти идентичных тРНК мы можем судить о относительном возрасте видов, родов (то о чем мы говорили выше)
davydov: одну секунду. мы кажется согласились с вами, что возраст всех видов один и тот же. незначительно различается скорость эволюции, и назвали это «консервативностью вида».
я: Да, это то что мы имеем сейчас… но судить мы можем о том, как они эволюционировали. Мы согласились с вами с тем, что мы сейчас взяли штаммы разных видов и сейчас их секвенировали, но не то что все виды появились в один момент. Так же?
davydov: ну, все виды являются потомками luca, какой момент мы будем брать за точку отсчета появления вида?
я: Мы это можем сделать лишь относительно, и судить об этом лишь на основании той информации которая у нас есть, поэтому все будет зависеть от того множества штаммов, которое мы рассматриваем. Тогда имея скажем 1000 штаммов нам надо их выстроить в виде последовательного временного развития. Начальная точка отсчета у нас появится скорее всего только в самом конце исследования. Вначале мы сможем лишь разделить эти 1000 штаммов на группы и не сможем сказать жили ли они параллельно или нет. Но те которые будут внутри группы будет означать, что они произошли где-то в одну эпоху
davydov: дело в том, что 1000 современных штаммов — это результат следующего процесса:
был один вид, он разделился на два, каждый из двух разделился ещё на два, …, и так 10 раз, в результате получаем 1024 штаммов. Все промежуточные значения утеряны, вы видите 1024 вида, а все предыдущие этапы вам неизвестны, потому что эволюция идет непрерывно.
я: это не совсем так, тут нам и помогут идентичные тРНК. Как отдельный вопрос.
davydov: просто вы пытаетесь найти промежуточные состояния, а среди современных штаммов их нет.
я: как же нет 😉 есть и это видно по консервативности
davydov: откуда? для них эволюция остановилась? Консервативности отдельных молекул, тогда как в других молекулах накопились отличия.
я: Ну, мы же уже говорили о этом
davydov: но видимо не до конца друг друга поняли. это ключевой вопрос. можно я приведу хорошую модель?
davydov: смотрите, у нас есть листовка. на ней напечатан текст. мы просим машинисток постоянно перепечатывать эту листовку на печатной машинке, а старые листовки при этом уничтожаем. машинистки довольно часто ошибаются, есть места, где ошибиться легко, есть места, где ошибиться сложно (консервативные и вариабельные участки). некоторые листовки вообще выбраковывают и не делают с них копий. мы даем одну листовку, приходим через год, у них на столе 1000 листовок с немного разным текстом. где-то длиннее, где-то короче и т.п. промежуточных листовок среди них нет.я: ок, и что мы разве не можем вычислить последовательность замен. Можем. Задайте вопрос как? 🙂
davydov: мы можем для каждой группы листовок приблизительно оценить, когда была та (последняя) листовка, которая была прародителем этой группы. мы можем приблизительно восстановить предковые состояния для каждой группы.
мы можем дать оценку, что такое-то свойство появилось не позже чем тогда-то (но не точную оценку).
например, мы видим что все листовки начинаются со слова «Здравствуйте»
мы можем предположить, что первая листовка также начиналась со слова здравствуйте, но мы не знаем, что было за 10 листовок до первой. может она всегда начиналась со слова здравствуйте, а может быть это ошибка вкралась на одной из предыдущих типографий.я: Не так
я: итак у нас есть 1000 листовок. И мы можем выделить 5 групп.1. 250 листовок, со словами
«Здравствуй Миша, как дела? У меня все хорошо.»
2. «Здравствуй Миша, как дела? Я уезжаю.» — 150 листовок
3. «Здравствуй Миша, как дела? Мне скучно, но все хорошо.» 100 листовок
4. «Привет Миша, как дела? Мне скучно, но все хорошо.» 100 листовок
5. «Добрый день, Михаил, как дела? Я уезжаю.» 400 листовокВсе остальные слова в листовках сильно перемешены, написаны с ошибками и не дают точной информации. Дальше как можно выстроить эти группы по образованию во времени исходя из общих фраз? Попробуйте. Не ужели ни как?
davydov: ну, допустим мы используем алгоритм neighbour joining. мы определим меру близости для каждой пары групп
потом объединим две самые похожие. потом присоединим к ним третью. потом четвертую. потом пятую. то есть мы можем «найти последнего общего предка» для каждой группы.
я: Так в чем же проблема?
davydov: мы также можем предположить, что в последнем общем предке всех существующих листовок был вопрос «как дела». (но вполне возможно, что в первой его и не было)
я: и что, то чего нет мы не анализируем
davydov: мы не можем определить время, когда возникла фраза «мне скучно, но все хорошо». мы можем дать только оценку, что это произошло не позже чем какой-то момент. и точно неверно говорить, что 2 произошла от 3. потому что они оба произошли от кого-то другого. и эта листовка уже давно уничтожена.
я: Мы не можем определить время в абсолютных цифрах — возможно, хотя и это можно но тут мы выходим за область биоинформатики.
davydov: согласен.
я: Можно говорить, что 2 произошла от 5 в том смысле, что мы рассматриваем только две информативные фразы. и эти фразы произошли в таком порядке вначале как у 2 а потом исказились как у 5
davydov: это не происхождение вида. это происхождение фразы.
я: ок, но эти фразы содержаться в какой то определенной листовке
davydov: да. и мы можем сказать, что мол в предке (столько-то поколений назад) её вероятно не было, а сейчас она есть. и появилась она где-то на огромном отрезке из точки а в точку б.
я: хорошо, не вижу противоречий. думаю мы о одном разными словами
davydov: отлично. мы научились определять момент появления гена. но мы по-прежнему не понимаю, что такое момент появление вида.
современные виды — это точки на ветвях эволюции. иногда эти ветви раздваиваются, но никогда не останавливаются, и нельзя выбрать такой-то возраст и сказать, что протеобактерии произошли тогда-то. потому что ни одной современной протеобактери тогда не было, были какие-то другие её предки.
я: но фразы фиксируются на листовках, хотя изучаем мы конечно эволюцию не листовок как материального носителя, а фраз
когда мы говорим о моменте появления вида, мы подразумеваем, что когда то существовала такая-то листовка, которая есть прообраз текущей и в которой содержалась такая же фраза
davydov: поскольку в реальности текст очень большой, то выбор конкретной фразы очень сильно повлияет на результат.
и вы скажете, что давайте называть проетобактериями тех, у кого есть такие-то гены. а я скажу, нет, давайте выберем другие гены. и у нас получатся существенные различия в результатах.я: А я скажу давайте ориентироваться только на те фразы, которые вообще не изменялись для отдельных групп
и выводы на основании эти жестких фраз будут более строгие, чем на основании фраз которые подвержены изменениям
davydov: у нас опять проблемы с определением, что такое вид и «когда он возник»
в сущности, то, что вы говорите — это и есть последний общий предок. просто неправильно называть момент существование последнего общего предка — моментом появления вида или рода. потому что род или вид могли возникнуть раньше.davydov: значит наше противоречие состоит в следующем.
вы утверждаете, что можно по каким-то признакам определить некий род или вид, момент его появления.
я говорю, что если мы уже имеем современное представление о неком виде или роде, то мы можем взять всех представителей этого вида или рода и найти последнего общего предка (и допустим оценить время его жизни).
при этом вполне возможно, что род или вид возникли сильно раньше. но точно не позжея: что-то вы меня запутали… с одной стороны, вы говорите что нельзя определить момент появления вида, с другой стороны говорите, что можно найти предка и время его жизни. Так если мы знаем время жизни предка — то мы знаем что момент появления вида находится на отрезки времени жизни предка — и нет тут противоречий.
davydov: время возникновения вида и время существования общего предка — это разные понятие.
знаете определения вида? для обезьян например, это возможность давать здоровое потомство.
так вот, возьмем последнего общего предка всех людей. вполне вероятно, что этот предок не может спариваться с современным человеком и давать здоровое потомство. значит он относится к другому виду. (и этот вид давно вымер)
это именно время появление последнего общего (для всех людей) предка, а не время, когда человек стал человеком.
так что время появления вида — это очень тонкий и сложный биологический вопрос.
— я предлагаю вам другую формулировку задачи: введем функцию D(A, B), которая для видов A и B дает значение, которое тем больше, чем раньше разделились ветви эволюции видов A и B. То есть тем больше — чем раньше существовал последний общий (для A и B) предок. Последний — это важно, потому что первый общий предок, это всегда luca, и для любой пары видов будет одинаковое значение. задача — найти эту функцию.
я: задача упрощенная одной функции мало, но как первое приближение пойдет. И могу ввести эту функцию — это число совпадающих идентичных тРНК
davydov: отлично. давайте по-позже продолжим? мне кажется, мы уже очень близки к сути
я: ок
davydov: ок. теперь вопрос, как эту функцию выбрать. слушаю вас.
я: так вот, один из способ — я считаю прямой, это число функционально значищих идентичных последовательностей ДНК которые не мутируют от вида к виду, от рода к роду и тогда мы приходим к тому, о чем я говорил про тРНК — они как раз такие
все же остальные способы косвенные и не точные. ок?
davydov: значит, тут есть несколько тезисов. во-первых, я правильно понимаю, что вы утверждаете, что тРНК не мутируют?
я: нет, конечно они мутируют вообще, но они МОГУТ не мутировать от рода к роду
davydov: прекрасно. значит они всего лишь консервативны.
я: да, больше чем все остальное
davydov: не спорю. какую именно процедуру вы предлагаете на основе тРНК?
я: тот график, который я показывал в статье на хабре — показывает лишь число идентичных тРНК между родами.
davydov: давайте по пунктам. 1. вы берете ряд видов, интересующих вас, так?
я: я беру все штаммы, которые есть в NCBI (National Center for Biotechnology Information) в базе секвенированных штаммов (ncbi genomes). Формат файла там .fna. Есть еще другие файлы, которые указывают якобы где находятся тРНК — вот там есть проблемы, но я и устраняю.
davydov: каким образом?
я: Есть файл .frn — там имеется последовательность тРНК, которые имеются в данном организме по мнению ученных (скорее всего они пользуются бластом или чем то таким). Важно то, что у тРНК конец должен оканчиваться на CCA, я это перепроверяю, и отбрасываю все которые не имеют. Далее длина тРНК не может быть произвольной, она как правило находится в районе 70-100 нуклеотидов, меньшие и большие я тоже отбрасываю. Получаю примерно 20000 уникальных видов тРНК
davydov: (лучше «типов» тРНК, чтоб не путаться)
я: да
davydov: типом вы называете точную последовательность
я: потом проверяю по всем файлам .fna все 20000 тРНК — есть ли они там действительно и где. Да, точную — один в один
davydov: как вы это делаете? технически, поиск подстроки?
я: программно — да поиск подстроки
я: так вот потом группируем по родам и получаем граф, по этому графу можно судить о связях между родами. Этот граф зашумлен конечно и по хорошему можно разрядить эти связи
davydov: Во-первых, в чем проблема этого графа. хотя погодите. давайте с простого, я просто тРНК никогда не занимался, поэтому не знаю: сколько у одной бактерии разных тРНК?
я: 2-3 на каждую аминокислоту, т.е. порядка 60
davydov: правильно ли я понимаю, что существуют разные штаммы, а может и виды, у которых тРНК абсолютно идентичны?
я: таких очень мало — исключение из правила, и только в рамках одного вида
davydov: хорошо. полезно заметить, что для этих групп штаммов тРНК вообще не дает никакой информации.
я: кроме того, что они один вид 🙂
davydov: и нужно рассматривать более вариабельные последовательности. ок? ну иногда хочется смотреть и на штаммы, но редко.
davydov: дальше вопрос, существует ли пара видов, для которой вообще отсутствуют общие тРНК?
я: да, существуют. и их много, найти общие проблема, а не наоборот
davydov: значит, для них анализ тРНК такую функцию не даёт. это означает, что в условиях ограниченной выборки наша функция плохо работает на больших расстояниях.
я: на графе есть вершины, которые не связаны с остальными — да их можно отбросить, или рассматривать лишь отдельно от остальных, но там есть достаточно большой участок 70% родов которые так или иначе между собой связаны
davydov: хорошо. просто запомнить, как некоторое ограничения метода.
следующий вопрос: у нас есть три вида. У них такие тРНК
1: AAABBB
2: AAABBA
3: BBBAAA
несмотря на то, что 1 и 2 явно ближе, они будут казаться также далеки, как 3?я: я не знаю, это проблема тех кто рассматривает тРНК с мутациями — против чего я возражаю
davydov: ну вы согласны с тем, что 1 и 2 ближе?
я: да, 1 и 2 ближе
davydov: но ваш метод этого не покажет (это гипотетическая ситуация, я понимаю, что на самом деле там скорее всего будут и другие тРНК, которые между 1 и 2 могут совпасть)
я: да, не покажет…
я: но в том то и дело, что в варианте с мутациями — вы демонстрируете на вырожденном состоянии, но таких нет среди 2000 геномов, которые мы имеем
davydov: допустим, да.davydov: хорошо. есть ещё две вещи
одна из них состоит в том, что по-видимому, бывают процессы (изменяющие систему трансляции) которые приводят к резкому изменению тРНК, не обусловленную большим эволюционным расстоянием.
т.е. что-то меняется в рибосоме, и сразу кажется организмы намного дальше, чем они есть на самом деле (если смотреть по другим признакам) есть работа на эту тему, я вам, наверное, давал ссылку. тезис понятен?
я: «бывают процессы (изменяющие систему трансляции)» — это все равно, что поверить в революцию, а не в эволюциюdavydov: нет, ну понятно, что на самом деле происходит все не мгновенно.
davydov: итак, у нас есть ваш метод, у него есть определенные достоинства, определенные недостатки.
+ очень простой и воспроизводимый алгоритм.
+ маленькая вероятность ошибиться, практически невозможно не найти, очень
+ маленькая вероятность ошибиться. практически невозможно найти не то, почти всегда нужное находится. можно for fun ещё посмотреть blast’ом сколько вы реально упустили, но, думаю, немного.теперь минусы
— на совсем близких расстояниях ваш метод работает плохо, т.к. не видит отличий. (это экзотика, но надо это помнить)
— на совсем далеких расстояниях метод тоже в текущем виде не работает, т.к. связей вообще нет. это хуже. потому что такое иногда нужно.
— причем! даже если связи есть, но их мало, то метод работает, но плохо, потому что он не учитывает гомологию двух трнк, если они отличаются хотя бы на пару позиций.я: все так. ну с этим можно жить 🙂
iakov.davydov: с этим можно, жить.davydov:
— метод подвержен смещению в тех видах, где произошли какие-то изменения в рибосоме.
я: это отдельный вопрос, и еще не ясно, это плюс или минус
davydov: и последнее, ваша мера (или функция) отражает два типа эволюционной связи.
а) вертикальное родство
б) горизонтальное родство (речь про горизонтальный перенос генов)я: это тоже скорее плюс
я: Конечно, можно ориентироваться и на мутации, чтобы исправить мой метод. Но на мутации ориентироваться хуже на больших расстояниях — они становятся не ясны. Поэтому мой метод как раз для первичного определения дальностиdavydov: я считаю, что ваш метод не универсален, но может быть использован для двух вещей.
а) для поиска горизонтального переноса генов (не любых генов, к сожалению, а только тРНК)
б) для поиска событий резкого изменения аппарата трансляции
я: согласен
Пример нахождения предка среди современников
Чтобы можно было объять мы рассмотрим пример только на 5 родах:
1 Salmonella
2 Enterobacter
3 Citrobacter
4 Marinomonas
5 Oceanimonas
Если выделить их из общего графа, то мы получим:
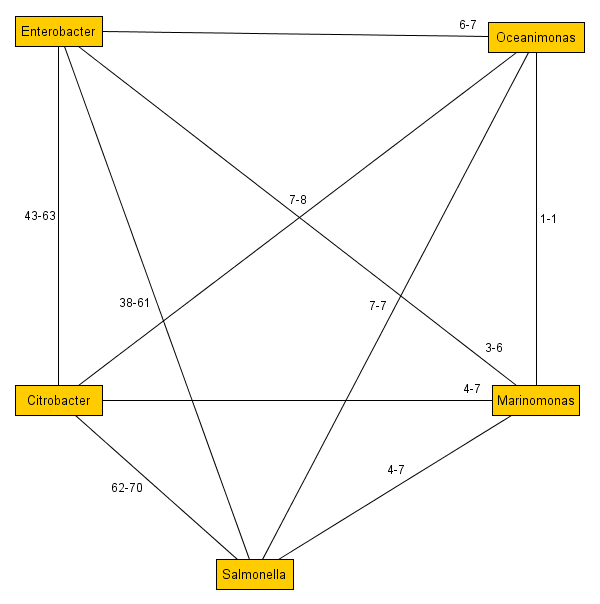
Видим, что тут каждая вершина связана со всеми остальными. Ни о каком направлении эволюции тут говорить не приходится пока. Граф не ориентированный и показывает лишь силу связей (минимум-максимум). Здесь видно лишь то, что Salmonella, Enterobacter и Citrobacter — связаны между собой более сильно. Но говорит ли это о эволюционной близости хоть что-то? В общем-то пока нет. Но классическая филогенетика с этим смиряется.
Но что мы можем сделать дополнительно. Мы можем проанализировать состав каждой связи. Каждая связь показывает минимум-максимум тРНК, которые идентичны между двумя родами. Но имея только лишь эти числа мы не видим состав этих связей. И по сути не можем судить, когда появился тот или иной род. Поэтому и говорим, что они братья. Но это не совсем так.
Первое что мы можем сделать, это предположить, что у родов Marinomonas и Oceanimonas был один предок X2, а у Salmonella, Enterobacter, Citrobacter другой предок X1. Кроме того, окажется, что X1 предок X2. (это все рассчитывается, но у еще не достаточно формализовано, поэтому лишь демонстрация, хотя алгоритм можно сделать). И тогда получится следующий ориентированный граф:

Цифорки на связях теперь означают состав идентичных тРНК, были выделены 5 групп:
1. группа из 7 тРНК
2. группа из других 7 тРНК
3. группа из 53 тРНК
4. группа из 6 тРНК
5. группа из других 6 тРНК
Числа из 6 цифер — это ID одного тРНК. В одном месте указано 1(-12804), что означает состав группы 1, но за исключением тРНК с ID= 012804. С плюсом это композиция групп и отдельных тРНК. И тогда мы видим, что если предположить, что у предка X1 имелись все 5 групп и несколько отдельных тРНК, то прослеживая по связям разницу видим как происходила дивергенция, причем с 100% точностью (т.е. детерминировано). (как это делится на группы, я опускаю, алгоритм когда будет пойдет в научную статью (если такое случится) )
А теперь поищем может у нас есть такой предок среди современников? Посмотрев внимательно увидим, что да есть — это Citrobacter. И можем тогда перерисовать.
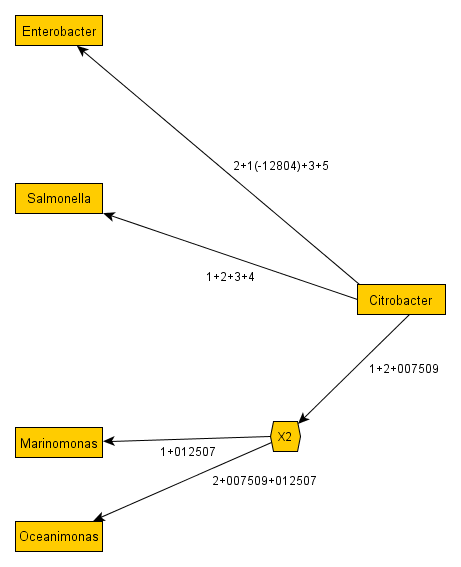
Так мы и нашли предка среди современников. (конечно, он предок не в полном смысле, но он тем не менее наиболее к нему близок по составу тРНК).
ссылка на оригинал статьи http://habrahabr.ru/post/164455/
Добавить комментарий